
Research
Section I — Vision and Theoretical Foundation
I-1. A Paradigm We Call "Drug-Minimal Medicine"
While advances in pharmaceuticals have dramatically extended the human lifespan, they have also fueled a negative spiral of "mass-production → mass‑administration → drug‑dependence.” Since 2013, the Taki Laboratory has pursued a vision we summarize as “One‑Shot, Side‑Effect‑Free, Drug‑Minimal or Drugless Society.” One of our objectives is to realize medicines that act for a prolonged period after a single dose and interact with nothing but the pathogenic target protein. One key lies in medium‑sized, covalent inhibitors we term biological Target Covalent Inhibitors (bioTCIs). (bioTI = reversible; bioTCI = irreversible/covalent.)
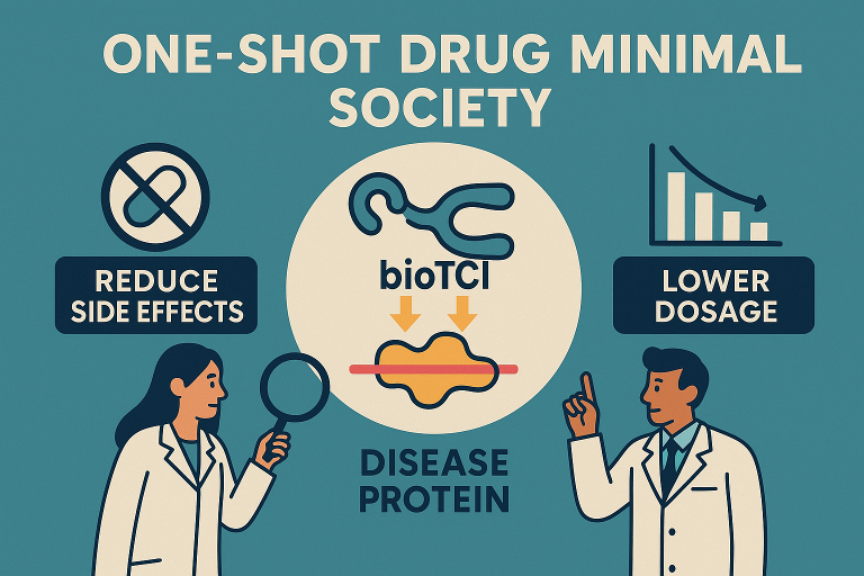
I‑2. Three Major Advantages of bioTCIs
・Long‑Lasting Efficacy — Because they form an irreversible covalent bond with the disease‑related protein, efficacy (PD) is sustained regardless of pharmacokinetic (PK) clearance.
・High Specificity & Low Side Effects — Peptides or nucleic acids, as “medium‑sized” scaffolds, engage the target protein through multivalent contacts, dramatically suppressing off‑target reactions that cause side effects.
・Flexible Chemical Design — Organic synthesis, evolutionary molecular engineering, and click chemistry can be freely combined for rapid, straightforward optimization.
FAQ: Why “Medium‑Sized” Molecules?
・Safety — Multivalent recognition of the target suppresses side effects.
・Synthetic Accessibility — Easy chemical/enzyme‑based modifications.
・Development Speed — Library technologies + high‑throughput selection (HTS) enable swift optimization.
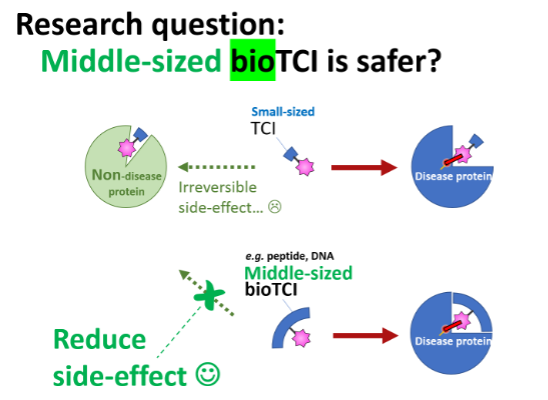
Challenges and Strategies
・Weaknesses of non‑covalent medium‑sized targeted inhibitors (bioTIs): susceptibility to proteases and rapid renal clearance.
・Weakness of conventional TCIs: permanent off‑target reactions caused by nonspecific binding of the small molecules.
Our laboratory focuses on technologies that overcome both issues:
1) After covalent binding, a bioTCI “self‑capsulates”, blocking proteases and gaining size to avoid renal filtration.
2) Before covalent binding, we combine half‑life‑extension chemistries such as Fc‑fusion.
3) During covalent binding, we deploy latent warheads that react strictly within “Match-Making Micro-Environment (MM-ME; ‘Trigger & Lock’ 2-step 2-step recognition mechanism) absent elsewhere in the human body.
I‑3. Layered Theoretical Infrastructure
| Layer | Laboratory Technologies & Orientation | Core Message |
|---|---|---|
| Chemistry | 10BASEd-T (10BT) on T7 phage, NEXT-A, Affinity/Reactivity Co-selection (ARC; ARC-10BT; ARCaDia) | Biocompatible yet highly tractable covalent reactions; Trigger & Lock‑type combinatorial selection. |
| Computation | bioTCI hub (NGS × MS × MD × AI) | Integrated learning of affinity + reactivity + dynamic structure |
| Measurement | Force-Responsive Materials/Device Co-invention | Real‑time integration of mechanical stimuli and pharmacological action. |
This multi‑layered architecture underpins a future in which “medicine is barely used at all.”
I‑4. Unresolved Scientific Questions — How Fundamental Science Opens Application
I‑4‑1. The Enigma of the Match‑Making Microenvironment (MM‑ME)Latent warheads such as aryl‑OSO₂F SuFEx are completely inert in aqueous solution, yet their reaction rate can increase by three orders of magnitude upon binding the target protein. This cannot be explained by simple proximity; we posit formation of an MM‑ME — a “zero‑turnover enzyme” that reshapes the electronic landscape of the latent electrophile via electrostatic fields, hydrogen‑bond networks, and desolvation. Sub‑picosecond dynamics, in which protein fluctuations and solvent rearrangements cooperatively propel the reaction coordinate, remain unobserved. Dissecting these micro‑reaction fields is key to making bioTCIs designable.
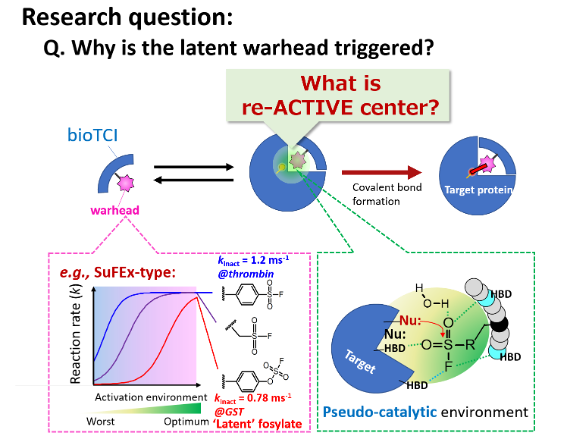
Question — Can we capture the moment the latent warhead awakens?
I‑4‑2. Essential Roles of Weak / Transient InteractionsThe first molecular encounter in vivo is an “encounter complex” (Kd µM–mM; τ µs–ms).
We hypothesize that this invisible step—akin to a tentative handshake—pre‑organizes the complex, matures the reaction field, and then cascades into irreversible covalent bonding.
By ‘freezing’ these weak / transient interactions using our ‘super-glue’, we aim to validate the true power of MM‑ME.
Weak/Transient Motif‑Finder — We integrate NGS (ARC enrichment), cross‑linking MS/MS (covalent sites), and coarse‑grained MD to computationally freeze µs‑scale complexes inaccessible to X‑ray/NMR.
AI proposes short core motifs (3–6 aa/nt) that feed back into new libraries.
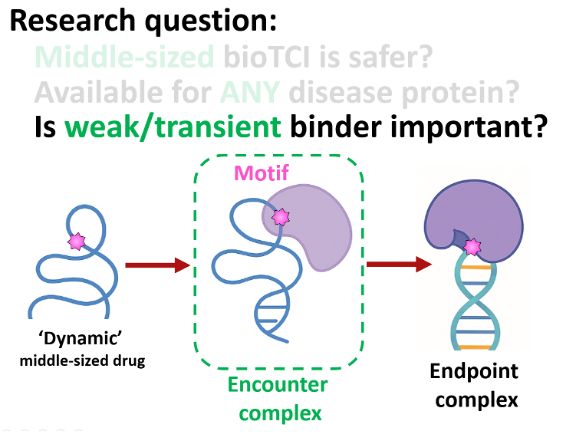
Question — Does biology start with a weak handshake, with the encounter complex cultivating MM‑ME?
Section II — Technological Concepts & Lab History
II‑1. Technology Timeline: Major Achievements & Platforms (2013–2025)
Key accomplishments of bioTCIs:We have established the combinatorial selection (ARC) platform for bioTCIs; because we are a small laboratory, collaboration with researchers of diverse expertise—including our university’s visiting professors—is indispensable.
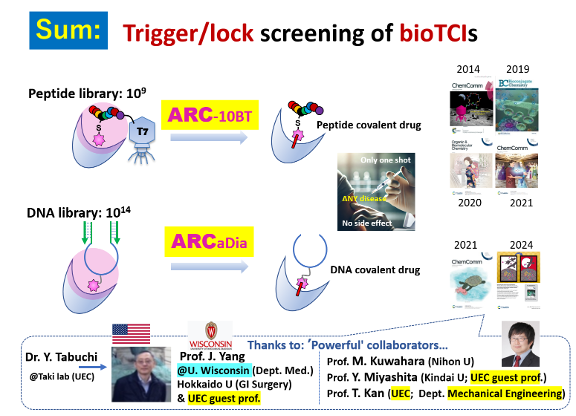
Our compact team is complemented by a network of cutting-edge professionals specializing in mass spectrometry, MD simulation, and mechano-optics, enabling multidisciplinary projects without the overhead of a large-scale lab.
Lab History Table:| Year | Highlight | Key Paper |
|---|---|---|
| 2013– | Discovered 10BASEd‑T (10BT) reaction enabling warhead installation onto T7 phage peptide libraries → start of bioTCI experiments Mol. BioSyst. 2013 (with Prof. Ito, Kagoshima Univ.; first author Dr. Fukunaga, now Assoc. Prof., Miyazaki Univ.) |
Mol. BioSyst. 2013 |
| 2018–19 |
1) First combinatorial selection of peptide‑type bioTCIs via indirect 10BT (mock warhead); direct 10BT still challenging BC, 2018 2) Precise Fc fusion into bioTIs via NEXT‑A (with Ajinomoto Co.) BC, 2019 (Front Cover) 3) First mechano‑gel whose fluorescence shifts with strain (seed of stress‑based therapy; with ETH Zürich & Keio Univ.) Soft Matter, 2019 (Back cover) |
1) BC, 2018 2) BC, 2019(Front Cover) 3) Soft Matter, 2019 (Back cover) |
| 2021 |
ARC‑10BT established: direct acquisition of peptide‑type bioTCIs via Trigger & Lock with latent warhead ChemComm, 2021 (Hot & Front Cover) |
ChemComm, 2021 (Hot & Front Cover) |
| 2024 |
ARCaDia: ultra‑rapid one‑round, non‑SELEX acquisition of DNA‑type bioTCIs (with UW & Hokkaido Univ. Med.) ChemComm, 2024 (Back cover) |
ChemComm, 2024 (Back cover) |
| 2025– | bioTCI hub (NGS×MS×AI reaction‑profile DB) and MechanoDB (3D stress‑imaging platform) launched Both peptide-type and DNA-type bioTCIs acquire serum-/nuclease-resistance once encapsulated |
— |
・Both peptide-type and DNA-type bioTCIs acquire serum-/nuclease-resistance once the covalent bond is formed.
・For DNA-type bioTCIs, administration of a complementary strand (CS) can neutralize the therapeutic effect on demand.
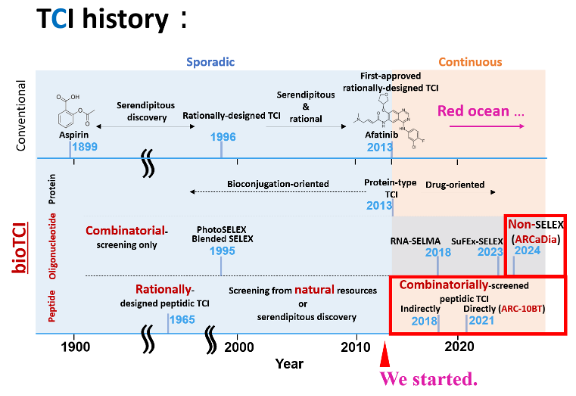
II-2. Our Laboratory’s Position in the (bio)TCI Research Timeline (red frame)
II‑3. Key Concepts Underpinning the Technology
II‑3‑1. Latent Warheads
Latent warheads such as aryl‑OSO₂F SuFEx (Sharpless et al. PNAS, 2018) are inert in water but activate exclusively within pseudo‑catalytic protein interiors. Such locale‑specific activation nearly eliminates side reactions and powers our ARC protocols to finish in a single round. No latent warhead yet combines dramatic rate acceleration with broad compatibility for diverse nucleophilic residues; designing one is a focus going forward.
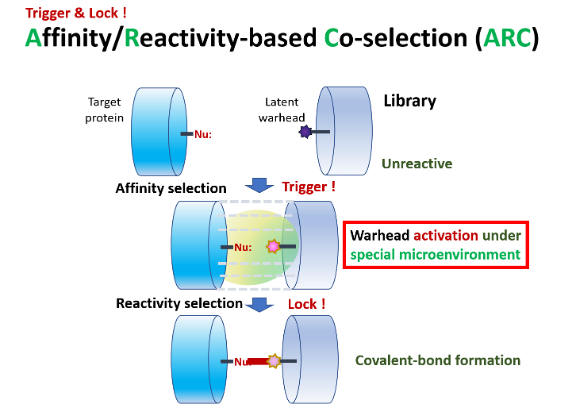
II‑3‑2. Affinity / Reactivity‑Based Co‑Selection (ARC); Trigger & Lock!
Step 1: target affinity “arrests” the binder and organizes the reaction micro-environment.
Step 2: reactivity “locks” via covalent bond — a two‑factor authentication (like smartphone) that eradicates side reactions.
Both peptide ARC‑10BT and nucleic‑acid ARCaDia also shorten timelines and simplify procedures.
Section III — Two Core Research Pillars (2025–)
bioTCI hub: Multilayer Data‑Integration Platform
True control of One‑Shot Therapy requires a quantitative model that formalizes the continuum of “weak binding → MM‑ME → covalent bond.” We will establish physical models of MM‑ME and a Weak/Transient Motif‑Finder:
・AI‑driven extraction of motif‑sequence–affinity–reactivity triplets from NGS big‑data, MS, and MD.
・A Reactivity‑AI module predicts reaction fields of new warheads.
・Free‑energy landscapes of ≥ tens of latent warhead–protein complexes reveal common “activation clusters.”
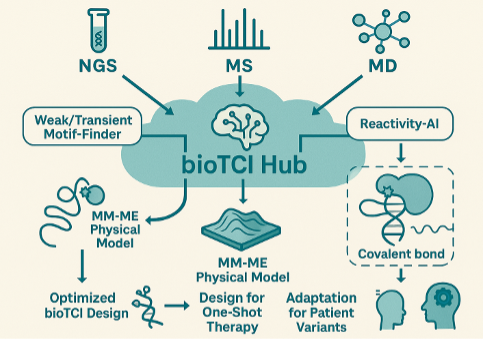
Outcome of bioTCI hub: mechanistic insights into MM‑ME and transient biomolecular interactions, bridging fundamental science and application.
Mechano‑3D Imaging & Force Therapy
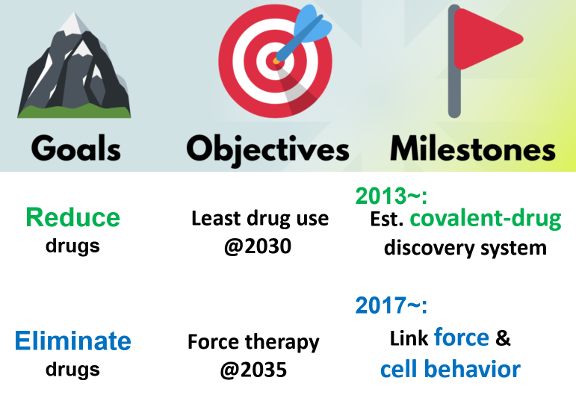
Co‑developing novel optical hydrogels and high‑sensitivity laser optics, we visualize three‑dimensional cellular stress fields in real time. Our goal is to correlate mechanical changes in cells (e.g., cancer malignancy, osteogenesis) with physiology at µm resolution, ultimately leading to “drug‑free therapy” (Force Therapy, 2035 – ) that manipulates cells in vivo/ex vivo.
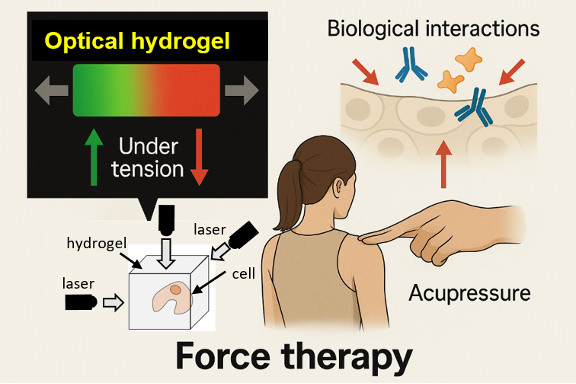
MechanoDB stores data for this composite materials / optics endeavor (joint work with Prof. Tomaru, NAOJ).